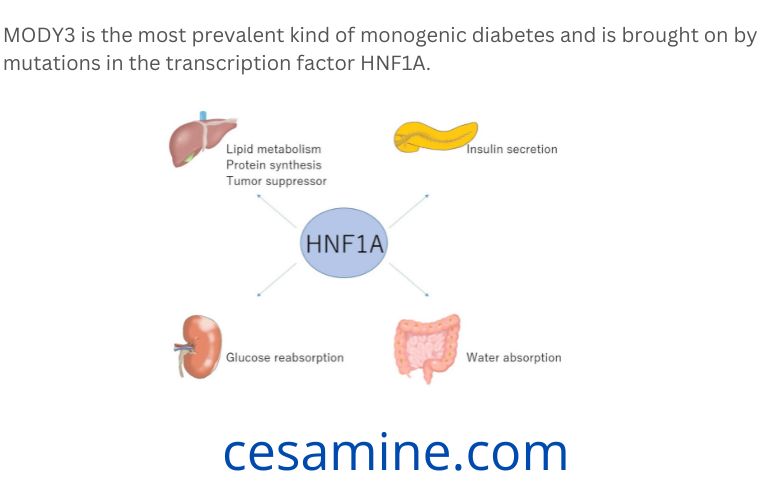
Understanding the disease mechanism is essential for providing diabetes patients with the best care possible. A genetic flaw in the HNF1A gene results in MODY type 3 (MODY3), a monogenic hereditary type of diabetes.
The end effect is progressive beta () cell degeneration, which triggers hyperglycemia, a disorder characterised by elevated blood sugar levels.
Why does MODY3 diabetes result from HNF1A mutations?
This issue was looked into by a research team under the direction of Henrik Semb, Director of the Institute of Translational Stem Cell Research (ITS) at the Helmholtz Diabetes Center of Helmholtz Munich. They discovered a new pathogenic mechanism for MODY3’s onset of diabetes. The journal Cell Stem Cell has officially published the findings.
There are various forms of diabetes from a medical perspective. One to two percent of instances of diabetes are due to the uncommon monogenetic (caused by the inheritance of a single gene mutation) condition known as maturity-onset diabetes of the young (MODY).
In the Caucasian population, MODY3 is the most prevalent kind of monogenic diabetes and is brought on by mutations in the transcription factor HNF1A. As a result of the cells’ disrupted insulin secretion, the patients’ blood sugar levels rise, causing hyperglycemia, which is a condition marked by elevated blood sugar levels. However, the aetiology, or how the illness arises, is still a mystery.
Researchers Henrik Semb and his group examined how MODY3 mice with HNF1A mutations develop diabetes over time using patient-derived stem cells. In MODY3, the researchers discovered a fresh pathogenic pathway underlying the onset of diabetes.
The MODY3 mutation under study led to insulin hypersecretion from beta cells. A significant discovery that will help keep those who carry the gene from developing diabetes.
In MODY3 cells, membrane depolarization is more effective.
The phenotypic of MODY3 patients is quite diverse, as seen, among other things, by the wide range in disease beginning ages. The factors that determine when a disease first manifests itself are poorly known, despite extensive research into the underlying illness process of MODY3.
A deeper comprehension of the causes of diabetes in MODY3 patients paves the way for focused therapies that postpone or even stop the illness.
First authors Florian Hermann and Maya Kjaergaard and their colleagues recapitulated the insulin secretion sensitivity to the membrane depolarizing drug sulfonylurea, a feature frequently found in MODY3 patients, using patient-specific induced pluripotent stem cells (iPSCs).
Unexpectedly, after being transplanted into mice, MODY3 patient-specific HNF1A+/R272C cells displayed an increased production of insulin both in vitro and in vivo. In comparison to their unaffected siblings, human HNF1A mutation carriers tend to be heavier at birth.
In MODY3 cells, decreased expression of potassium channels, particularly the KATP channel, led to an increase in calcium signalling. Additionally, the fact that the insulin hypersecretion phenotype may be reversed by pharmaceutically targeting ATP-sensitive potassium channels or low voltage-activated calcium channels suggests that the MODY3 cells’ hypersecretion of insulin is caused by more effective membrane depolarization.
Important knowledge to prevent or postpone the beginning of diabetes
The research emphasises the value of using patient-specific iPSCs as a platform for researching early disease pathways that open the door to personalised treatment.
The findings highlight the significance of early detection of hyperinsulinemia in people who carry the HNF1A mutation. Hyperinsulinemia is a condition in which the blood insulin concentration is unusually high. Reduced blood sugar levels result, and if they fall too low, they could be fatal.
With this information, Henrik Semb’s team of researchers opened the way for more research to determine if therapies to prevent hyperinsulinemia, including diets or medications, in neonates with HNF1A mutations will postpone or even prevent the emergence of MODY3 diabetes later in life.